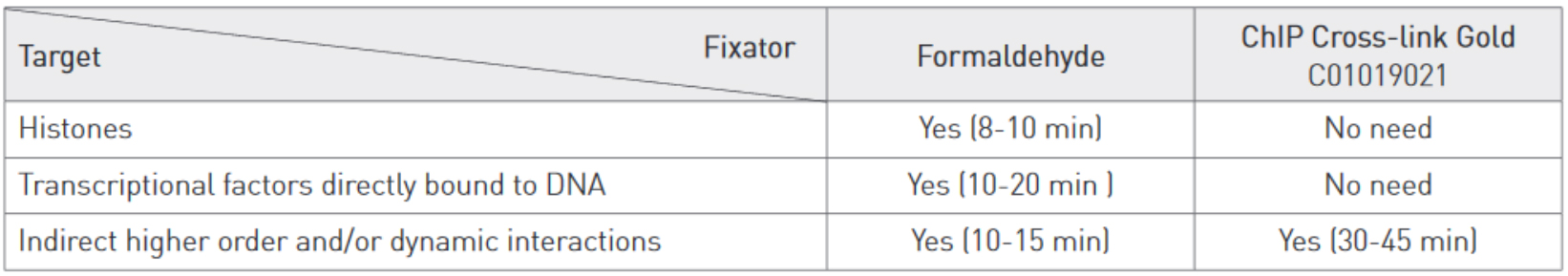
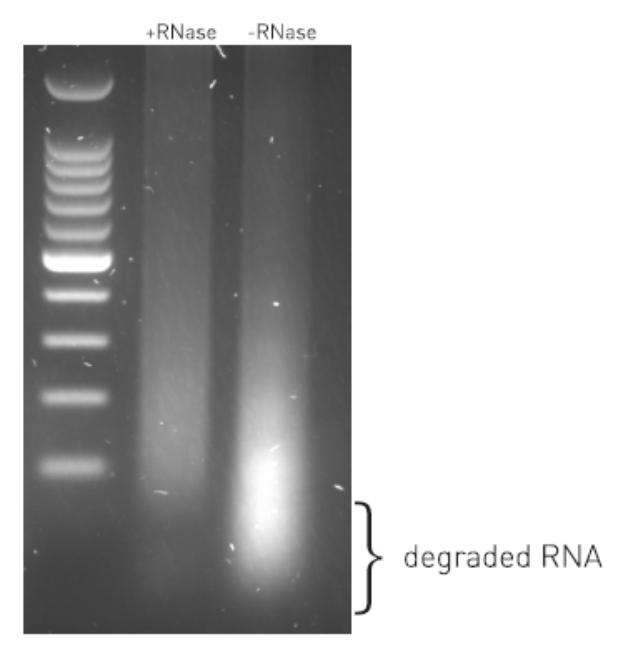
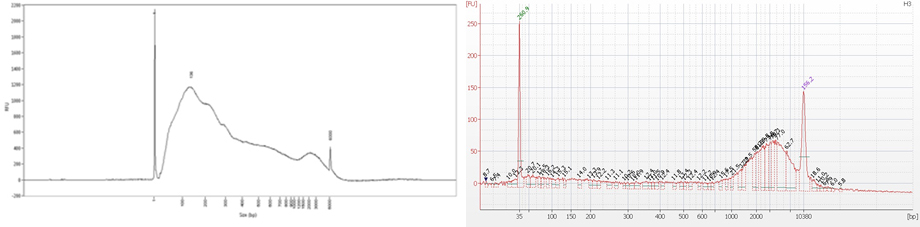
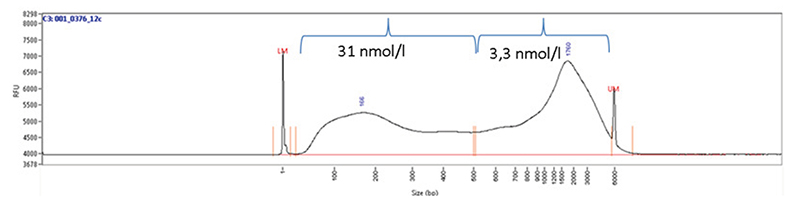
Cross-linking is typically achieved by using formaldehyde which forms reversible DNA-protein links. Formaldehyde rapidly permeates the cell membranes and enables a fast cross-linking of closely associated proteins in intact cells. Formaldehyde cross-linking is ideal for two molecules which interact directly. However, for higher order and/or dynamic interactions, other cross-linkers should be considered for efficient protein-protein stabilization such as Diagenode ChIP Cross-link Gold, an innovative dual cross-linking ChIP fixation reagent (Cat. No. C01019021).
Adopt the fixation strategy that is best suited to studying your particular proteins. Some epigenetic marks may be more elusive than others. When studying weak or rare protein-DNA binding events fixation should be done promptly and directly in medium. When studying histone marks, cells can be put in suspension by trypsinization before fixation. Generally, a shorter fixation is required for histones marks (8-10 min) than for transcriptional factors (10-20 min) using a standard formaldehyde single step fixation protocol.
Use fresh formaldehyde. The use of high quality and fresh formaldehyde is crucial while using methanol-free reagent is not mandatory. Replace your stock every month. This will ensure high inter-assay reproducibility between ChIP experiments.
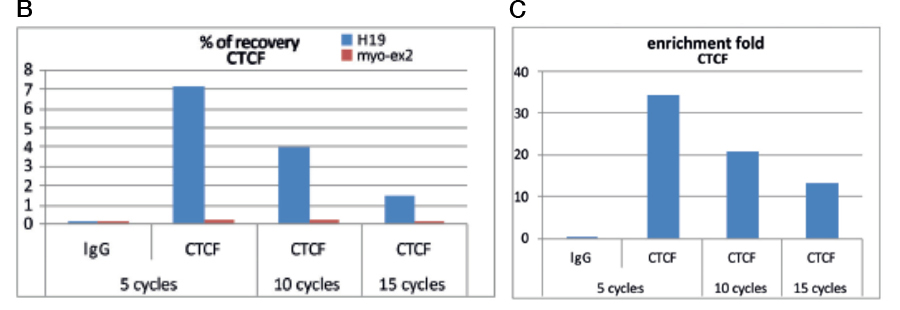
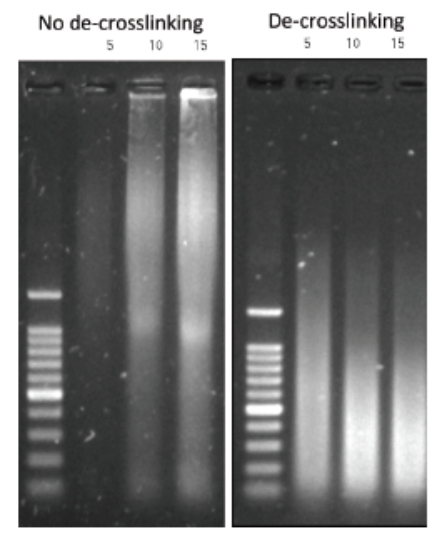
Always carry out a fixation time course for your cell line to empirically determine the optimal fixation time for your cell line and epitope of interest. Cell lines and epitopes differ widely in their fixation efficiency and sensitivity to fixation.
Be precise with the fixation time and temperature. The formaldehyde fixation is a time and temperature–depending process. A stronger cross-linking will be achieved at a higher temperature and at longer duration. Whether you use RT or 37°C, 5 or 15 min (depending on your specific target and cell types), make sure that the temperature and time are consistent.