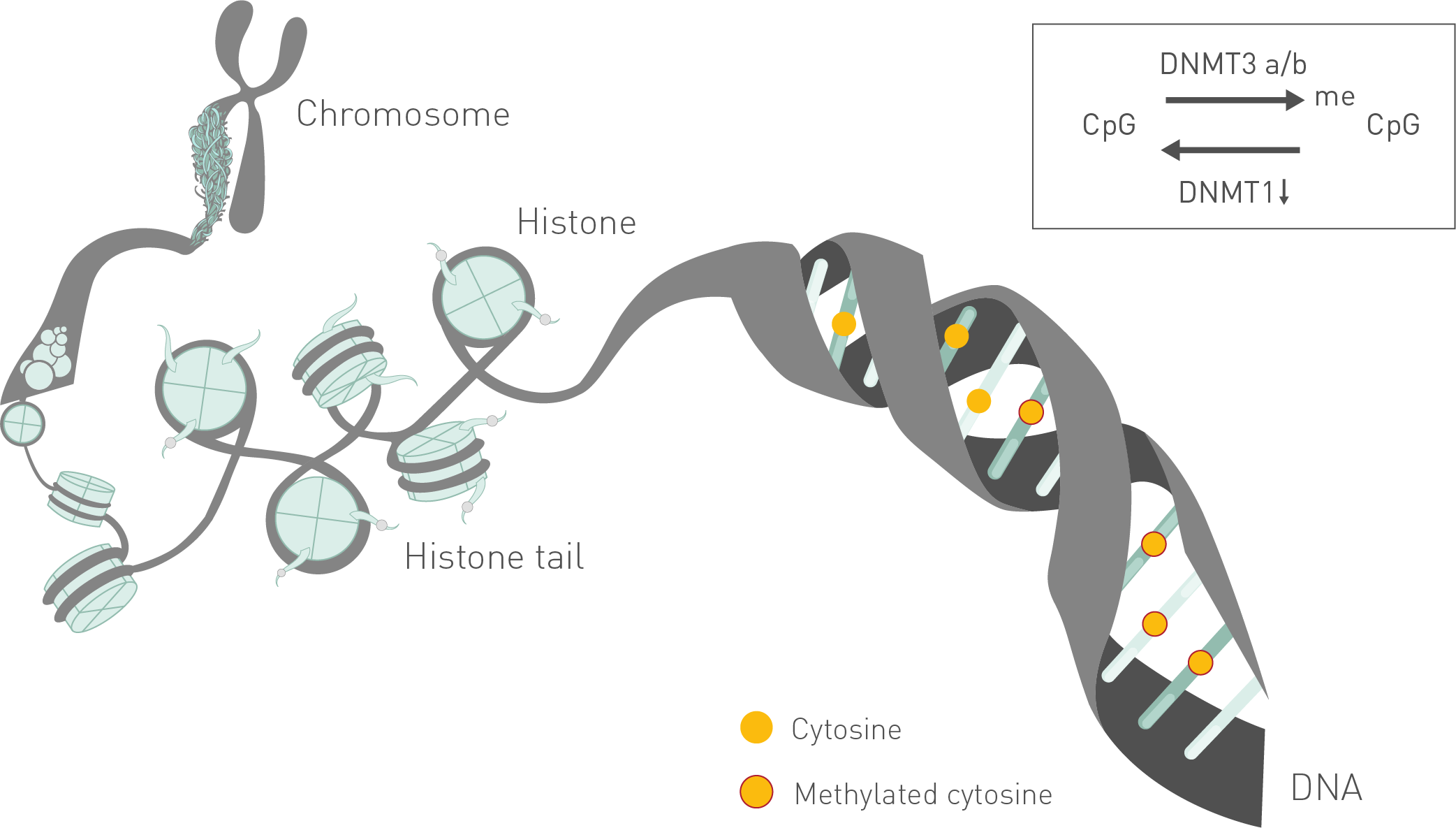
This analysis provides information on each single CpG with its methylation percentage.
Standard Analysis:
Summary statistics (total sequenced reads, total mapping reads, uniquely aligned reads, PCR duplicates (WGBS), number of CpGs detected, average coverage at CpG sites, number of CpGs detected with coverage greater than 10x, etc.)
Trimmed and filtered reads in fastQ files after sequencing QC
BAM sorted files from alignment to reference genome (indexed bam files and bigwig files included)
BED files from methylation calling and extraction (CpG location, number of methylated cytosines, number of unmethylated cytosines and coverage at the CpG site)
Advanced Analysis
Comparative analysis (also called differential analysis) aimed at finding differentially methylated CpGs (DMCs) and differentially methylated regions (DMRs) between two groups of samples
Annotation of DMCs and DMRs for genomic regions (exons, introns, etc) and for CpG island location (islands, shores, shelves, etc)
Gene ontology enrichment analysis on genes associated with DMCs and DMRs
Pathway enrichment analysis on genes associated with DMCs and DMRs (KEGG or DOSE for human samples)