News
Automated MethylCap, a validated method for high throughput genome wide DNA methylation profiling.
November 15, 2012
Researchers have extensively reported that methylated regions identified by MethylCap-seq could be validated with classical bisulfite sequencing (1 to 4).
Recently, a paper was published reporting a medium scale study on a collection of 24 colorectal cancer sample using Methyl-cap with Diagenode’s IP-Star automated workstation (5). In this assay, 2687 frequently hypermethylated and 468 frequently hypomethylated regions were identified. These data were then confirmed by the bisulfite conversion method.
Methylation of CpG dinucleotides is generally associated with epigenetic silencing of transcription and is maintained through cellular division. Multiple CpG sequences are rare in mammalian genomes, but frequently occur at the transcriptional start site of active genes, with most clusters of promoter CpGs being hypomethylated.
The Diagenode H6-GST-MBD fusion protein of the MethylCap kit has been extensively validated. It consists of the methyl binding domain (MBD) of human MeCP2, as a C-terminal fusion with Glutathione-S-transferase (GST) containing an N-terminal His6-tag. The H6-GST-MBD fusion protein can be used to specifically isolate DNA containing methylated CpGs. See overview below.
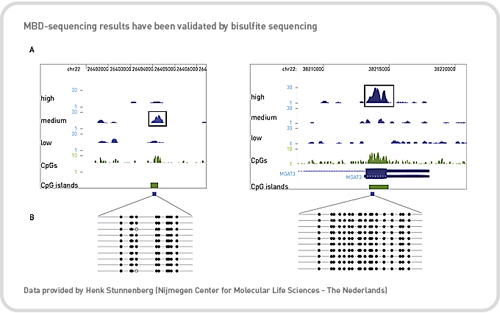
Figure 1: Using the MethylCap approach, two methylated regions were detected in different elution fractions according to their methylated CpG density (A). Low, Medium and High refer to the sequenced DNA from different elution fractions with increasing salt concentration. Methylated patterns of these two different methylated regions were validated by bisulfite conversion assay (B).
References
- Brinkman AB, Simmer F, Ma K, Kaan A, Zhu J, Stunnenberg HG. Whole-genome DNA methylation profiling using MethylCap-seq. Methods 2010; 52:232-6; PMID: 20542119; DOI: 10.1016/j.ymeth.2010.06.012.
- Bock C, Tomazou EM, Brinkman AB, Müller F, Simmer F, Gu H, et al. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol 2010; 28:1106-14; PMID: 20852634; DOI: 10.1038/nbt.1681.
- Bogdanovic O, Long SW, van Heeringen SJ, Brinkman AB, Gómez-Skarmeta JL, Stunnenberg HG, et al. Temporal uncoupling of the DNA methylome and transcriptional repression during embryogenesis. Genome Res 2011; 21:1313-27; PMID: 21636662; DOI: 10.1101/gr.114843.110.
- Brinkman AB, Gu H, Bartels SJJ, Zhang Y, Matarese F, Simmer F, et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res 2012; 22:1128-38; PMID: 22466170; DOI: 10.1101/gr.133728.111.
- Simmer F, Brinkman AB, Assenov Y, Matarese F, Kaan A, Sabatino L, Villanueva A, Huertas D, Esteller M, Lengauer T, Bock C, Colantuoni V, Altucci L, Stunnenberg HG. Comparative genome-wide DNA methylation analysis of colorectal tumor and matched normal tissues. Epigenetics. 2012 Oct 18;7(12).