How to properly cite our product/service in your workWe strongly recommend using this: クロマチンデータ解析 (Hologic Diagenode Cat# G02010107). Click here to copy to clipboard. Using our products or services in your publication? Let us know! |
クロマチンデータ解析
Catalog Number
Format
G02010107
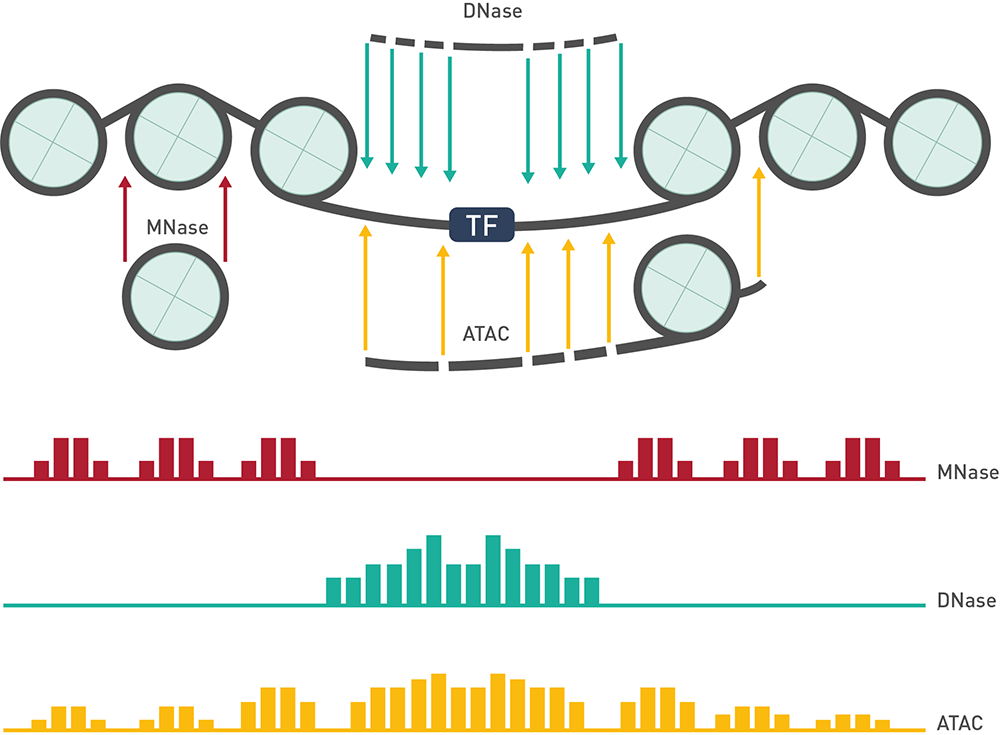
クロマチン免疫沈降(ChIp-Seq)とそれに続くATAC-Seqは、エピジェネティックな規制研究に使用される2つの一般的な技術です。 ChIP-Seqは、ヒストンを含む特定のタンパク質が結合する遺伝子制御領域にかかわるオープンクロマチン領域の解析を可能にし、ATAC-Seqは、遺伝子制御領域に関するオープンクロマチン領域を解析に役立ちます。 Diagenodeでは、結合部位とアクセス可能なクロマチン領域を正確に検出するために、最適化されたコンピューティングパラメーターを採用。お客様のプロジェクトに合わせて、特定のパラメータをカスタマイズして頂けます。
目的のタンパク質やプロジェクトによって、大きく異なるシーケンス深度、リード幅、ペアリングモードを含め、詳しくは当社テクニカルサポートにご相談ください。
解析から何がわかりますか
この解析でわかるのが、標的タンパク質が特異的に結合する各領域(ピーク)と、その濃縮レベル(ChIP-seq)または開放性(ATAC-seq)に関する情報です。
標準解析
- Summary statistics (total sequenced reads, total mapping reads, uniquely aligned reads, PCR duplicates, number of peaks, average peak width, reads in peaks, frequency of reads in peaks, etc.)
- Trimmed and filtered reads in fastQ files after sequencing QC
- BAM sorted files from alignment to reference genome (indexed bam files and bigwig files included)
- BED files from peak calling (peak location, peak calling score, peak density)
- Annotation files with location of the peaks within genes
高度解析
- Comparative analysis (also called differential analysis) aimed at finding differentially bound sites or uniquely bound sites between two groups of samples
- Annotation of differentially/uniquely bound sites
- Determination of specific binding motifs within peaks
- Gene ontology enrichment analysis on differentially binding/unique sites
- Pathway enrichment analysis (KEGG or DOSE for human samples)
カスタマイズ解析
If you require a type of analysis that is not in the previous list, please consult with our expert bioinformatics team.
ChIP-seq
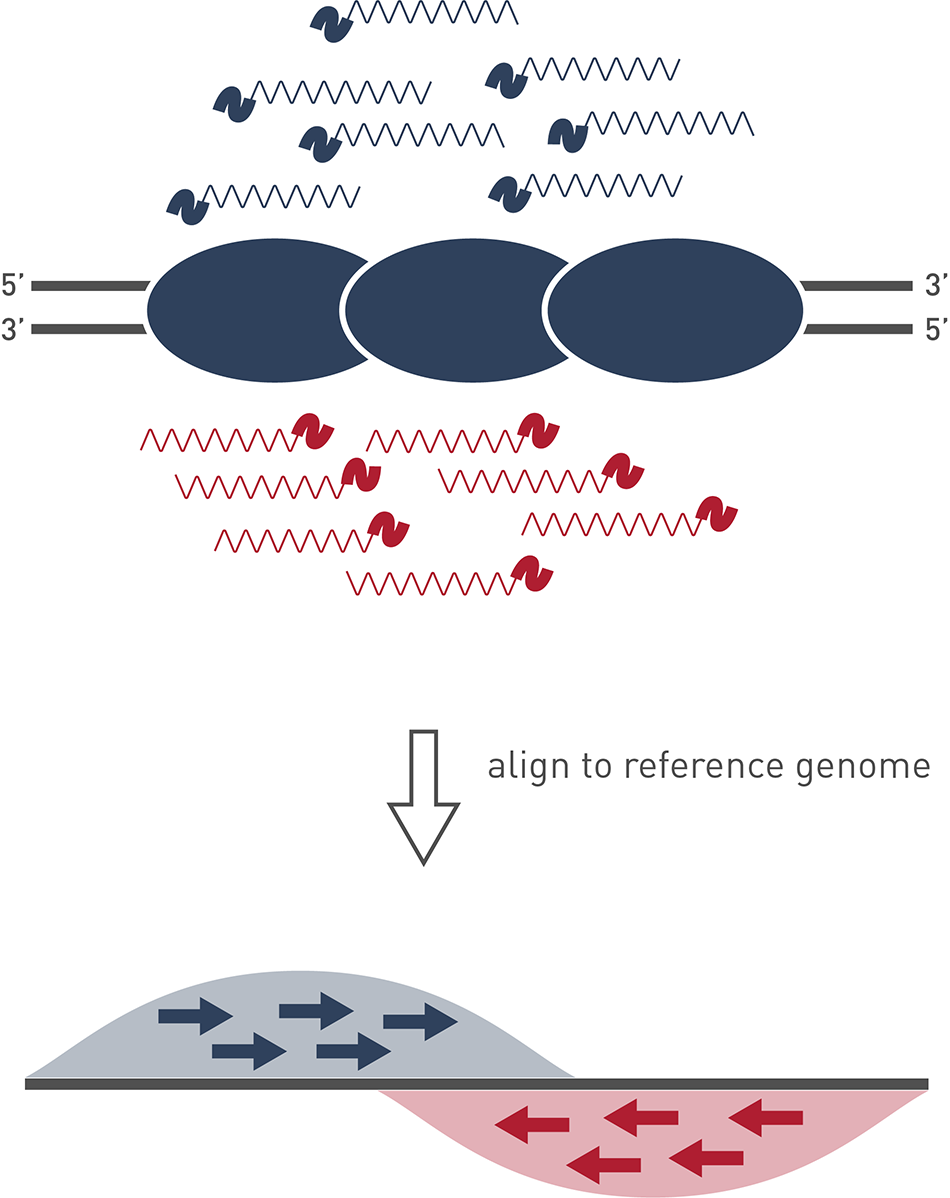
ATAC-Seq